Key Mechanisms
Some General Ideas
While blanket generalities can often be dangerous, there are some ideas that can be used systematically to help us become comfortable with mechanism, especially in the second semester where the pathways become more detailed and more complicated. Knowing the main concepts behind Substitution, Elimination, and Addition from Organic 1 in detail will make Organic 2 that much more manageable.
Mechanisms will either be concerted or stepwise, they will involve reversible and/or irreversible processes, and they will be governed by the ideas of steric environment and/or electronic stability. There aren’t many options here so it is important to understand that the paths open to molecules in chemical reactions are fairly limited.
Know About Conditions
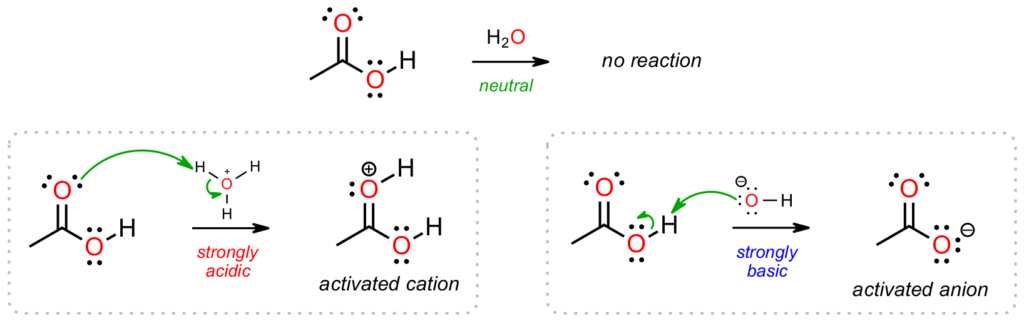
For “neutral” conditions in a reaction read inert or unreactive first since the substrate itself is unlikely to be attacked as there are no strong acids or bases present. The substrate will have time to “do its own thing” for example by breaking apart in carbocation-based reactions.
Consider the SN1 reaction between a tertiary alkyl halide in an alcohol solvent. The pH of the mixture will be neutral when the chemicals are first introduced so there is no strong acid to be attacked or strong base to do the attacking. The alkyl halide is able to collapse to a carbocation, which then attracts the electron-rich O of the alcohol and an ether is formed. In the example below, the stable carboxylic acid does not react with water (or alcohols) at neutral pH as there is nothing that reactive present.
Environment Matters
If we can identify the conditions in a reaction as being acidic (sulfuric acid, HBr, HCl, etc.) we already know what is going to happen first; the organic substrate is going to attack the acid and the substrate will become activated. This is of major importance in acid-catalyzed reactions where neutral conditions are not sufficient for reactivity but adding an acid gets things going by activating the organic substrate. This shows up in dozens of mechanisms in Organic 1 and 2 and gives an element of predictability to how pathways unfold. Since we are dealing with a “positive” environment, any intermediates formed during these pathways will be positively charged (oxonium ions, carbocations, etc.).
In the acidic environment below left the substrate attacks the acid to yield a cationic oxonium ion intermediate, which may react further depending on what else is present. The environment in the below right example has changed to strongly basic so the “attacker” and “attackee” change. Now the negative (electron-rich) base can attack the (electron-poor) carboxylic acid proton as a way to become stable through deprotonation. Notice how the mechanism arrows have changed direction in going from acidic to basic conditions. In general, organic substrates attack acids while bases attack organic substrates. This is a reliable indicator of what will happen in other mechanisms in the different pH environments.
SN2
Substitution Bimolecular — Concerted
Consider the reaction between the secondary alkyl bromide shown and sodium cyanide in DMF as a polar, aprotic solvent. We know that this gives the inverted product of substitution in which the cyanide anion has replaced the bromide. We also expect the new C-C bond to be stronger than the C-Br bond that is lost. So, what clues lead to the 2 arrow mechanism shown below?
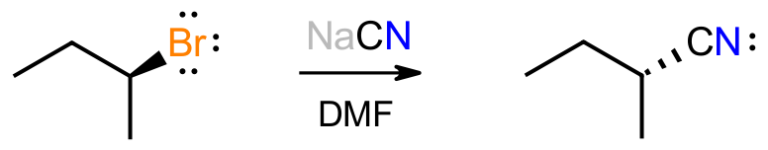
Working out the Arrows
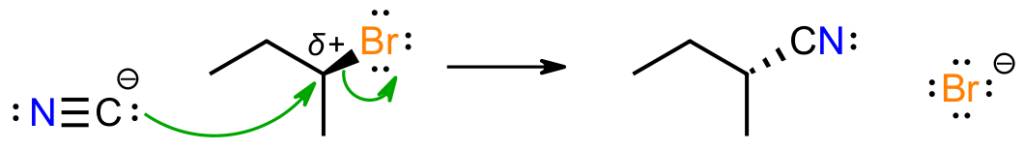
Firstly, with HCN having a pKa of about 9.2, the reaction is basic because of the cyanide salt, which suggests that the basic cyanide will attack the organic substrate. Since there are no obviously acidic protons on the substrate, there won’t be an acid-base reaction, and the negative cyanide will be attracted to the electron-poor alpha carbon attached to Br.
We only have two choices for how this mechanism plays out; either everything happens at once in a concerted pathway, or the bromine breaks off first in a stepwise pathway to give a carbocation, which then reacts with cyanide to give product. The second pathway would give enantiomeric products, i.e. a racemic mixture, because a flat carbocation would be formed. This does not happen so we focus on the concerted path.
The mechanism shown matches the paradigm described previously. The basic reagent attacks the substrate, from the back where the anti-bond is, with the bromine being displaced to maintain the octet at carbon. The negative charge has now been transferred from carbon to bromine, which is more electronegative and bigger so handles negative charge better. This mechanism matches the experimental evidence, in which the stereochemistry is inverted, and the kinetic data that shows the reaction to be bimolecular.
SN1
Substitution Unimolecular — Stepwise
Now we look at the reaction between a chiral tertiary alkyl bromide and methanol to give a racemic mixture of ethers. The pH of the reaction starts out as neutral, meaning there are no strong acids or bases present to react with the substrate. The alcohol is also not a strong nucleophile although the tertiary carbon is too crowded to be attacked anyway. Since we know that the Br is replaced, it has to break off at some point, so how does that happen to fit the observed racemic outcome?

Working out the Arrows
In the SN2 pathway the Br was kicked out in a concerted process, here it must break off first, as part of a stepwise pathway, which agrees with the observed rate-determining step being unimolecular. That would generate a flat carbocation, which in this case would be prochiral and accessible from either face. Rather than the alcohol “attacking” the cation, we could think of the cation “recruiting” the weakly nucleophilic alcohol, which would be close by as it likely serves as the solvent in this solvolysis process.
Equal attachment from either side would lead to a racemic mixture of protonated ethers, which would then lose the extra proton to the solvent to furnish the ether products. The mechanism fits the observed data, and also the idea of assessing what can or will happen based on reaction pH. Here the neutral conditions meant the alkyl halide was not attacked and had time to collapse to the carbocation.

E2
Elimination Bimolecular — Concerted
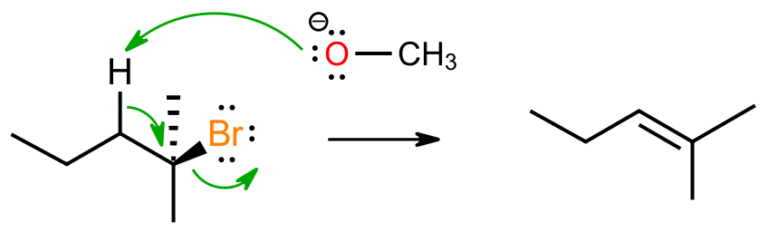
When we move to a hindered substrate under strongly basic conditions, we see a change in pathway to bimolecular elimination, i.e. the E2. Strongly basic conditions means the base attacks the substrate, however the SN2 is impossible as the alpa carbon is too crowded for approach. The base, here sodium methoxide, is trying to become stable, by transferring its extra lone pair (i.e. the negative charge) to the bromine leaving group, which can handle it better. Deprotonation at the beta carbon allows for a logical flow of electron density to form an alkene.
Smaller bases tend to give the more stable, more highly substituted Zaitsev alkene, while larger bases give the less substituted Hofmann outcome. Not only is this a useful method in synthesis, but it also tells us that the elimination is irreversible since establishment of an equilibrium, if the reaction was reversible, would give the thermodynamically, more stable, Zaitsev product. The outcome must therefore depend on transition state energies and be kinetically driven.

Working out the Arrows
We know that basic conditions won’t allow stepwise carbocation formation, and that the reaction here in base has a bimolecular rate-determining step, so the reaction must be concerted. It is possible for all required bonds to be formed and broken at once, so a concerted pathway is the most logical option. Regioselectivity is important here but stereoselectivity is also observed with the trans isomer often being favoured, again due to preferred geometries in the possible transition states.
E1
Elimination Unimolecular — Stepwise
When the conditions change to acidic, alkyl halides are mostly inert, but alcohols will react to give alkenes, especially at higher temperatures. The Zaitsev regioisomer is usually favoured and the rate-determining step is unimolecular. Higher temperatures serve two purposes. Firstly, this boosts the entropic factor (T delta S) in the Gibbs equation, and secondly, this allows the thermodynamically unfavourable alkenes to be distilled out of the equilibrium as they are formed. This method serves as a simple and reliable protocol for producing Zaitsev alkenes that takes advantage of the process being reversible.
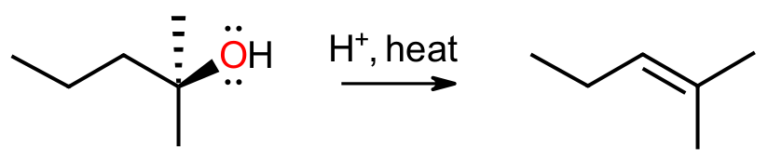
Working out the Arrows
To come up with a mechanism, following the established rules means the substrate, here the alcohol, attacks the acid to generate a hydronium leaving group. Since we are not in basic medium, the leaving group is able to break off in the unimolecular rate-determining step to give the carbocation. This species may then react with water as nucleophile to go back to the alcohol, or the water may serve as a weak base and deprotonate to give the alkene.
This whole process is stepwise and most likely an equilibrium. The more stable alcohol will actually be favoured, since sigma bonds are generally stronger than pi bonds, but the alkene can be the major product by removing it from the mixture as it is formed. This will then be replaced in the equilibrium and result in a high-yielding synthesis of the alkene.

HX Addition
Electrophilic Addition — Stepwise
When alkenes are treated with acids such as HCl, HBr, or HI they undergo addition reactions to give saturated alkyl halides with the more highly substituted products being favored. The acidic conditions mean the alkene substrate will attack the acid and that carbocations are viable as intermediates. Kinetic studies show that the protonation step is rate-determining and, when prochiral substrates are used, the products are formed as racemic mixtures.
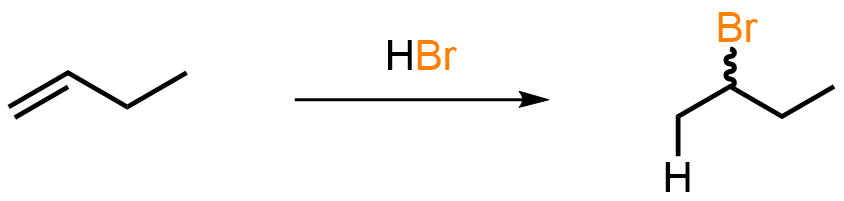
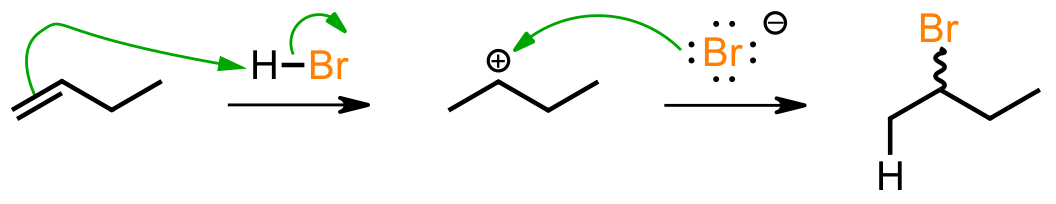
Working out the Arrows
The evidence suggests that electrophilic addition of HBr to the alkene involves two steps and goes through carbocations. The first step is formation of the carbocation in which the pi electrons from the alkene attack the hydrogen atom of HBr, which results in the positively charged carbon and a bromide ion. The stability of the resulting carbocation is significant and more stable carbocations such as secondary and tertiary are favored. The second step is then nucleophilic attack in which the bromide ion acts as a nucleophile and attacks the positively charged carbon. This results in the formation of the final product, the alkyl bromide.
The regioselectivity of the reaction is determined by Markovnikov’s rule, which translates to: when HBr adds to an unsymmetrical alkene, the major product is formed via the more stable carbocation intermediate. If the carbocation happens to be prochiral, as in this case, a racemic mixture of enantiomers is formed.
Borane Addition
Borane Addition — Concerted
The addition of a borane to an unsymmetrical alkene, followed by an oxidative workup with basic peroxide, results in the isolation of a racemic mixture of alcohols. Addition of the borane occurs with anti-Markovnikov regioselectivity and the stereochemistry of the addition is retained in the second step. Both regio- and stereochemical outcomes give clues to the mechanism involved.

Working out the Arrows
While the 6-electron boron is Lewis acidic the B to H bond is not polarized with boron having an electronegativity of 2.0 and H 2.1. This will mean that the pi bond in the alkene will not be protonated but will attack the electron-poor boron instead. Addition of the boron to the less substituted end of the alkene suggests that the regioselectivity is governed more by steric factors than electronic. Also, formation of only two stereoisomers and not four is evidence for a concerted syn addition and not stepwise through a carbocation. Since stereochemistry is retained in the second step that too must be concerted since stepwise formation of a carbocation would give more stereoisomers.
The observed regiochemistry and stereochemistry here leads to the conclusion that addition occurs through a concerted pathway, and the second, oxidation, step must also involve a concerted migration. While the borane reagent is Lewis acidic it is not a protic acid so carbocations are unlikely. Since the second operation is carried out under basic conditions a carbocation cannot be involved.

Summary
How do these foundational mechanisms help?
There is a common misconception that Organic changes every day and that there are no foundational rules in the subject. This isn’t the case of course. Knowing something about pH gives a solid idea of what is and is not going to happen in a mechanism. Strongly acidic will mean that a substrate is getting protonated while strongly basic means a substrate will be attacked. Neutral or acidic allows for carbocation formation but base generally does not. If a reaction gives only certain stereoisomers as products it probably means that flat intermediates such as radicals or carbocations are not involved since they can be attacked from two directions.
Stereochemical Clues
Compare each of the three concerted mechanisms discussed in the examples above and note that the stereochemical outcomes give clues as to how the changes occured. Notice that they all give certain stereochemistries but not all possibilties since no intermediate is formed. Conversely, the stepwise processes of SN1, E1, and electrophilic addition each give racemic mixtures of enantiomeric products since carbocations are formed and are attacked from two directions.
Regiochemical Clues
Regiochemical clues are also helpful, for example in the E2 reaction in which the size of the base can dictate which regioisomer will be formed as the major product. Small bases favor more stable Zaitsev outcomes while big bases generally give less-substituted Hofmann alkenes. The fact that the more stable products are not formed in the latter is strong evidence for the E2 process with base not being reversible and not equilibrating since that would give the Zaitsev outcome each time.